Securing a European Union Marketing Authorisation for a veterinary medicinal product is a highly regulated process, designed to ensure that animal medicines meet rigorous standards of safety, efficacy, and quality. Introduced in 2022, the Centralised Procedure simplifies this process, enabling pharmaceutical companies to obtain approval across the European Economic Area with a single application. A fundamental, yet often neglected, aspect of this process is the Linguistic Review of Product Information translations coordinated by the European Medicines Agency. This article explores the key stages of reviewing and approving Product Information in 24 European languages for centrally authorised veterinary medicinal products, offering practical guidance to help pharmaceutical companies navigate this critical step with confidence.
What Is the Marketing Authorisation Process for Veterinary Medicines in the EU?
The authorisation of veterinary medicinal products in the European Economic Area (EEA), also known as the European Single Market, is a tightly regulated process aimed at ensuring that treatments for animals meet strict safety, efficacy, and quality standards. Beyond protecting animal health, the framework also serves to safeguard public health and minimise environmental impact.
There are four routes available, depending on the product’s characteristics, intended use, and geographical scope:
- National Procedure – Limited to a single Member State, and typically used for veterinary medicines targeting local needs, such as region-specific diseases or species.
- Mutual Recognition Procedure (MRP) – Extends an existing authorisation in one Member State to others within the EEA.
- Decentralised Procedure (DCP) – Used when simultaneous authorisation is sought in multiple Member States.
- Centralised Procedure (CP) – Provides a single application for veterinary medicines requiring approval across all EEA Member States.
What Is the EU Centralised Procedure for Veterinary Medicinal Products?
The Centralised Procedure (CP), established under Regulation (EU) No 2019/6 (effective from 28 January 2022) and managed by the European Medicines Agency (EMA), provides a streamlined pathway that enables companies to submit a single application for evaluation and authorisation.
Veterinary medicines eligible for the CP fall under mandatory or optional scopes. The mandatory scope includes products developed using specific biotechnological processes (e.g., recombinant DNA technology, monoclonal antibodies or gene expression techniques), novel therapies, performance-enhancing products, and biopharmaceuticals containing engineered tissues. Generics, hybrids, and other products not included in the mandatory scope, can be assessed through the CP provided that no prior Marketing Authorisation (MA) exists within the EEA.
Following a thorough evaluation, EMA’s Committee for Medicinal Products for Veterinary Use (CVMP) issues a Positive Opinion. The European Commission then grants final approval, allowing the product to be marketed across the EEA.
What Is the Product Information?
The Product Information (PI) is a set of documentation that communicates essential details about the product. It comprises three annexes, each serving a specific purpose:
- Annex I: Summary of Product Characteristics (SmPC or SPC)
Provides key technical details, including active ingredients, target species, indications, dosage, and contraindications. It is primarily intended for veterinary professionals to support them with making informed treatment decisions. - Annex II
Outlines conditions of use or specific requirements for the product’s MA, if there are any.
- Annex III: Labelling and Package Leaflet (PIL)
- Labelling: Contains text for immediate or outer packaging (e.g., blister packs, bottle labels, cartons)
- PIL: Written in lay terms for end-users, such as veterinary nurses, livestock caretakers, or pet owners and included with the product supplied for sale.
- Labelling: Contains text for immediate or outer packaging (e.g., blister packs, bottle labels, cartons)
- PIL: Written in lay terms for end-users, such as veterinary nurses, livestock caretakers, or pet owners and included with the product supplied for sale.
What Is the EMA Linguistic Review of Product Information?
EMA’s Linguistic Review of the PI is an essential component of the CP, ensuring the accuracy, consistency, and quality of the English PI (EN PI) and its translations into 24 EU-EEA languages. Although the process for veterinary medicines differs slightly from that for centrally authorised human medicines — and sometimes uses different terminology for similar actions — the fundamental principles remain the same. It starts with the submission of the EN PI, which undergoes detailed evaluation and revision. Once finalised, the EN PI is translated into 24 EU-EEA languages. The 24 language PIs (also known as national texts) are reviewed by national competent authorities in respective EU-EEA Member States to ensure compliance with regulatory requirements before final approval.
The review process is divided into two stages, each with strict timelines measured in calendar days.
Pre-Opinion Review
The pre-opinion review applies to all new Marketing Authorisation Applications (MAAs) and certain Variations Requiring Assessment (VRAs). Conducted in two phases, it starts on Day 0 with the submission of the EN PI to EMA.
Phase One: Initial Evaluation
By Day 10, EMA evaluates the EN PI against its Quality Review of Documents (QRD) template and conventions to ensure compliance with formatting, layout, and content guidelines. Feedback from EMA, rapporteurs, and the CVMP is consolidated during review milestones at Days 85 and 120 into a single EN PI file. This document is then sent to the applicant as part of the CVMP’s List of Questions (LoQ).
Phase Two: EU-EEA Member State Review
At Day 121, the applicant returns the revised EN PI to EMA, which distributes it to relevant EU-EEA Member States for further comments. Member States primarily focus on QRD compliance and provide feedback by Day 140. Additional comments from rapporteurs and the CVMP are integrated at Days 150 and 180. The applicant must address all comments and provide the updated EN PI by Day 181. Any further feedback from rapporteurs must be resolved by Day 190 to enable the CVMP to adopt its Opinion on Day 210.
Post-Opinion Review
The issuance of a CVMP Positive Opinion marks the start of the post-opinion review process on Day +0, shifting focus exclusively to the 24 EU-EEA language PIs.
For Initial MAAs
By Day +5, the applicant provides translations of the adopted final EN PI, along with QRD Form 1, via Eudralink. EMA distributes these to the EU-EEA Member States for detailed review. The Member States check the translations for quality and compliance with QRD guidelines, returning their comments and feedback directly to the applicant by Day +19. The applicant implements the required changes and submits the finalised package by Day +25, which includes:
- 25 annotated Word PIs (24 languages plus English),
- 25 clean Word and PDF PIs (24 languages plus English),
- QRD Form 2
- INN table, if applicable
By Day +27, EMA verifies that all comments have been addressed and transmits the finalised files to the European Commission. Between Days +29 and +39, the Standing Committee conducts its consultation, and the European Commission issues its legally binding Decision by Day +55.
For Generic and Hybrid Products
The post-opinion review also applies to generic and hybrid products (i.e., containing the same active substance and used at the same dose(s) to treat the same condition as their reference product). Since the EN PI should, in principle, be very similar to that of the reference product, a pre-opinion review is not required.
By Day +5, the applicant submits to EMA the adopted EN PI, the 24 language PIs, and an annotated EN PI showing differences from the reference product’s EN PI. Subsequent steps follow the same timelines and process as for initial MAAs.
For Post-Authorisation Procedures
Post-authorisation procedures involve variations to the terms of an MA that may affect the PI. These variations are classified by their need for scientific assessment and the extent of their impact on the PI.
Variations Not Requiring Assessment
In rare cases where the variation does not require scientific assessment (VNRAs) but affects the PI, the Marketing Authorisation Holder (MAH) provides annotated EN and 24 language PIs as part of the variation application. No further review is conducted.
Variations Requiring Assessment
For variations requiring scientific assessment (VRAs), the impact on the EN PI determines the applicable review procedure. VRAs are further classified into three categories based on their assessment timelines:
- Reduced timetable (VRA-R)
- Standard timetable (VRA-S), such as G.I.18 variation
- Extended timetable (VRA-E)
If the EN PI is affected, the MAH provides clean and annotated Word versions of the EN PI showing all proposed amendments as part of the variation application.
Handling Changes to the EN PI
The type and extent of changes to the EN PI dictate whether a pre-opinion review, a post-opinion review, or no review is required:
Minor Changes
No post-opinion review is necessary. By Day +5, the MAH submits the updated 24 language PIs directly to EMA, and no further review by Member States is conducted.
Major changes
Only a post-opinion review is required. EMA informs the MAH of the timelines when the CVMP Positive Opinion is communicated. By Day +5, the MAH submits the 24 language PIs with the adopted amendments tracked directly to EU-EEA Member States. Their review focuses solely on these amendments.
Substantial changes
Both pre-opinion and post-opinion reviews are conducted, following the same process and timelines as for initial MAAs.
Grouping Variations
To streamline the process and reduce administrative burdens, MAHs can group multiple variations affecting the same MA into a single submission.
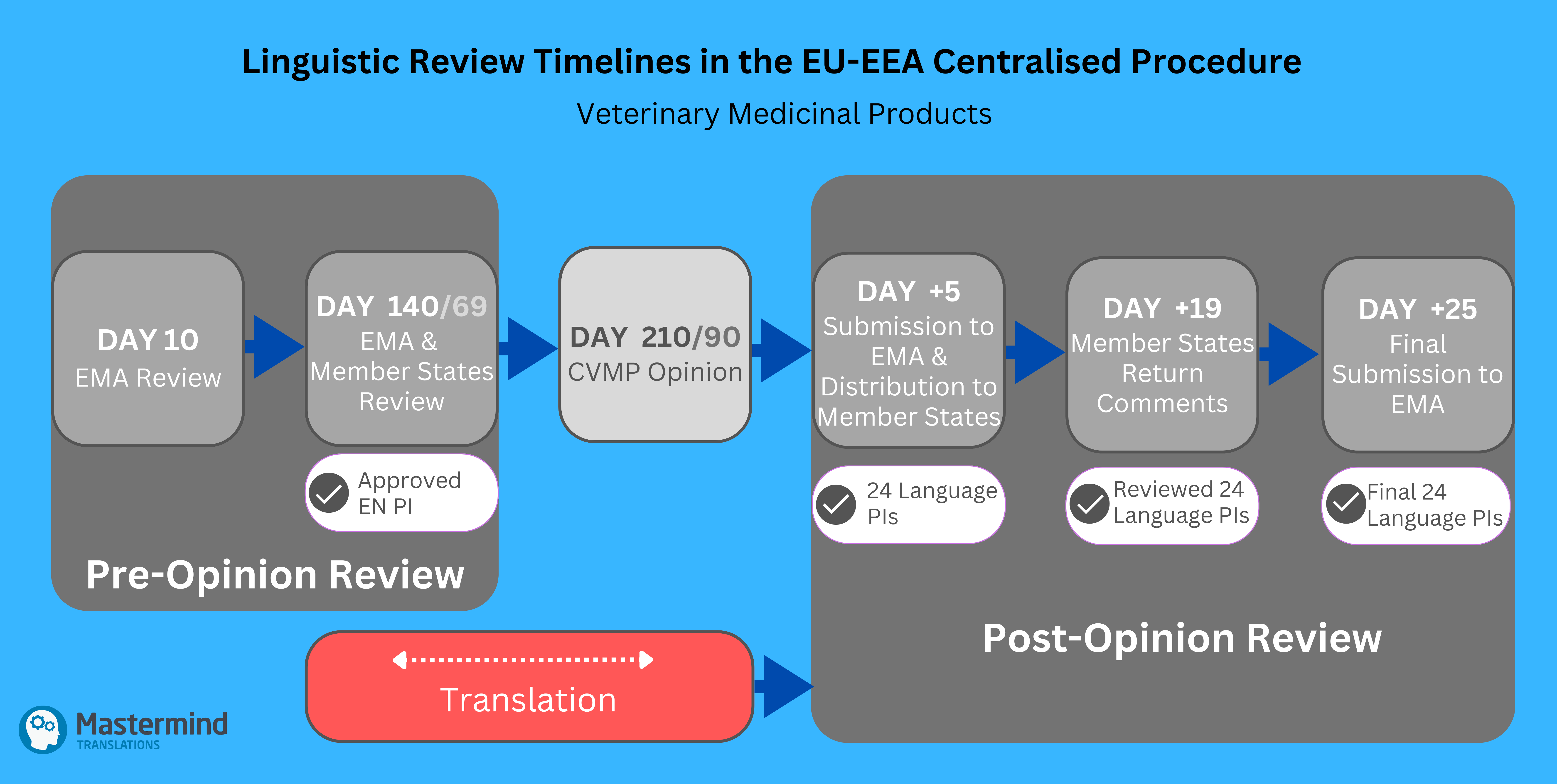
What Are the Key Timelines And Their Implications for the Translation Process?
- Day +5 Submission
Once the CVMP Positive Opinion is issued, the 24 language PIs must be provided within 5 calendar days. Given the short timeframe, and the complexity and scope of work, MAHs are strongly advised to initiate the translation process well in advance during the Pre-Opinion stage. This is particularly important for initial MAAs and variations with substantial changes, such as alignment with QRD Product-Information Template 9.0. Ideally, the Day 121/180 version of the EN PI should be shared with the translation provider to allow them sufficient time to prepare the translations, which can then be finalised after the last update at Day 0. - Day +25 Submission
After Member States return feedback on Day +19, MAHs have only 6 calendar days to incorporate comments and finalise the language PIs. The final package, including annotated and clean Word versions of the 24 language PIs, must be submitted by Day +25.
Best Practices for QRD Veterinary Translations in the EMA Linguistic Review Process
To ensure an efficient, compliant translation process, follow these best practices:
- Partner with an Experienced Language Service Provider
Select an LSP with proven expertise in veterinary regulatory affairs and specific experience in managing translations for centrally authorised veterinary products. A specialist life science translation service provider will be better equipped to understand the complexities of your project and have a pool of vetted translators who are competent in veterinary translation. - Communicate Requirements and Expectations Clearly
Provide your LSP with a detailed overview of your product’s MA procedure and its current stage. This clarity will enable the LSP to fully understand your specific requirements and critical time points, allowing them to design a tailored solution aligned with your objectives. This is particularly important for variations requiring substantial updates, such as QRD alignment, and grouped variations. Working closely with your LSP will ensure that all translations meet the expectations of national authorities responsible for reviewing the 24 language PIs. - Start Translations Early
Where applicable, initiate the translation work before the CVMP Positive Opinion. Sharing a draft EN PI during the pre-opinion stage ensures adequate preparation time, reducing errors and avoiding unnecessary delays in the post-opinion review. - Use a Phased Approach
Support your LSP by implementing updates in waves rather than all at once. This structured strategy allows for thorough quality assurance checks and ensures compliance in accordance with EMA’s fixed deadlines. - Maintain a Locked EN PI Version
Avoid unnecessary changes to the EN PI once shared with your LSP. Even minor adjustments can disrupt translation workflows, complicate version control, and delay completion.